La Holoprosencefalia
La holoprosencefalia es un trastorno cefálica en el que el prosencéfalo (el cerebro anterior del embrión) falla a desarrollar en dos hemisferios. Normalmente, se forma el cerebro anterior y la cara comienza a desarrollarse en la quinta y sexta semana de embarazo. La condición también se produce en otras especies.
La afección puede ser leve o severa. En la mayoría de los casos de holoprosencefalia, las malformaciones son tan graves que los bebés mueren antes de nacer.
Cuando el cerebro anterior del embrión no se divide para formar bilaterales hemisferios cerebrales (las mitades derecho e izquierdo del cerebro), que causa defectos en el desarrollo de la cara y en la estructura y función del cerebro.
En los casos menos graves, los bebés nacen con normal o casi normal desarrollo del cerebro y de las deformidades faciales que pueden afectar los ojos, la nariz y el labio superior.
Síntomas
Los síntomas de la holoprosencefalia desde leve (sin defectos faciales / órganos, anosmia, o sólo una única central de los incisivos) a moderada a grave (ciclopia).
Hay tres clasificaciones de holoprosencefalia.
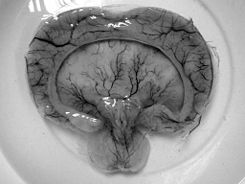
Patología macroscópica muestra de un caso de alobar holoprosencefalia.
- Alobar holoprosencefalia, la forma más grave, en la que el cerebro no es capaz de separar, por lo general se asocia con anomalías faciales severas, incluida la falta de una nariz y los ojos se fusionaron en una sola estructura mediana, ver Cyclopia
- Semilobar holoprosencefalia, en el que los hemisferios del cerebro han dividido un poco, es una forma intermedia de la enfermedad.
- Lobar holoprosencefalia, en la que existe una considerable evidencia de hemisferios cerebrales separados, es la forma menos severa. En algunos casos de holoprosencefalia lobar, el cerebro del paciente puede ser casi normal.
- Syntelencephaly, o variante interhemisférica media de la holoprosencefalia (MIHV), en el que la parte posterior del lóbulo frontal y el lóbulo parietal no se separan adecuadamente, pero el Rostro basales del cerebro anterior separa adecuadamente; es posible que esto no es una variante de HPE en absoluto, pero actualmente está clasificado como tal.
Presentación
Holoprosencefalia consiste en un espectro de defectos o malformaciones del cerebro y la cara. En el extremo más grave de este espectro se encuentran los casos de graves malformaciones del cerebro, malformaciones tan graves que suelen causar aborto involuntario o muerte fetal. En el otro extremo del espectro están los individuos con defectos faciales que pueden afectar los ojos, la nariz, y la parte superior del labio – y el desarrollo cerebral normal o casi normal. Las convulsiones y retraso mental puede ocurrir.
El más grave de los defectos faciales (o anomalías ) es ciclopía, una anomalía que se caracteriza por el desarrollo de un solo ojo, situado en la zona normalmente ocupada por la raíz de la nariz, y una nariz que falta o una nariz en forma de un trompa (un apéndice tubular) situado por encima del ojo. La condición también se conoce como cyclocephaly o synophthalmia, y es muy raro.
Causas
La causa exacta (s) de HPE aún no se han determinado, aunque se puede sospechar la presencia de toxinas. Sonic Hedgehog Gene, que participan en el desarrollo del sistema nervioso central, puede tener una mutación y causar Holoprosencefalia (USMLE Primeros Auxilios 2014). Sin embargo, a menudo parece que no existe una causa específica en absoluto.
Genética
Armand Marie Leroi describe la causa de ciclopia como un mal funcionamiento genético durante el proceso por el cual el embrión cerebro está dividido en dos. [ 4 ] Sólo más tarde la corteza visual tome forma reconocible, y en este punto un individuo con un solo cerebro anterior región ser propensos a tener un solo, posiblemente bastante grande, ojo (en un momento tal, los individuos con hemisferios cerebrales separadas se forman dos ojos)
Los aumentos en la expresión de tales genes como Pax-2, así como la inhibición de Pax-6, de la notocorda se han implicado en la diferenciación normal de las estructuras de la línea media cefálicos. Expresión inapropiada de cualquiera de estos genes puede dar lugar a formas de leves a severas de la holoprosencefalia. Otros genes candidatos han sido localizados, incluyendo el SHH (holoprosencefalia tipo 3 también conocido como HPE3), TGIF, ZIC2 y Six3 genes.
Aunque muchos niños con holoprosencefalia tienen normales cromosomas, específicos anomalías cromosómicas se han identificado en algunos pacientes (trisomía del cromosoma 13, también conocido como el síndrome de Patau). Existe evidencia de que en algunas familias, HPE se hereda (autosómica dominante y autosómica o recesiva ligada al cromosoma X herencia). Características compatibles con la transmisión familiar de la enfermedad (por ejemplo, un único centro incisivo superior) deben ser cuidadosamente evaluados en los padres y miembros de la familia.
Factores no genéticos
Numerosos posibles factores de riesgo han sido identificados, incluyendo la diabetes gestacional, infección transplacentaria (el “complejo TORCH”), primer sangrado trimestre, y una historia de aborto involuntario. Además, el trastorno se encontró el doble de frecuencia en el sexo femenino bebés. Sin embargo, no parece haber correlación entre HPE y la edad materna.
Hay evidencia de una correlación entre HPE y el uso de diversas drogas clasificadas como potencialmente peligroso para embarazadas y madres lactantes de madres. Estos incluyen la insulina, las píldoras anticonceptivas, la aspirina, litio, thorazine, ácido retinoico y los anticonvulsivos. También hay una correlación entre el alcohol y el consumo de HPE, junto con la nicotina, las toxinas en los cigarrillos y las toxinas presentes en el humo del cigarrillo cuando se utilizan durante el embarazo).
Pronóstico
HPE no es una condición en la que el cerebro se deteriora con el tiempo. Aunque los trastornos graves de convulsiones, disfunción autonómica, complicados endocrinos trastornos y otras afecciones potencialmente mortales a veces pueden estar asociados con HPE, la mera presencia de HPE no significa que se producirán o desarrollarse con el tiempo estos graves problemas, sin ninguna indicación o advertencia previa. Estas anomalías se reconocerán poco después de nacer o recién nacidos y sólo se producen si se fusionan las áreas del cerebro que controla las funciones, malformados o ausentes.
El pronóstico depende del grado de fusión y la malformación del cerebro, así como otras complicaciones de salud que pueden estar presentes.
Las formas más graves de encefalopatía suelen ser mortales. Este trastorno consiste en un espectro de defectos, malformaciones y anomalías asociadas. Discapacidad se basa en el grado en el que se ve afectado el cerebro. Moderado a defectos graves pueden causar retraso mental, espástica tetraparesia, atetoides movimientos, trastornos endocrinos, la epilepsia y otras enfermedades graves; defectos cerebrales leves sólo pueden causar problemas de aprendizaje o de comportamiento con pocas deficiencias motoras.
Las convulsiones pueden desarrollar con el tiempo, con el riesgo más alto antes de los 2 años de edad y el inicio de la pubertad. La mayoría son gestionados con un medicamento o una combinación de medicamentos. Por lo general, las convulsiones que son difíciles de controlar aparecen poco después del nacimiento, lo que requiere combinaciones de medicamentos / dosis más agresivas.
La mayoría de los niños con HPE están en riesgo de haber elevado en la sangre niveles de sodio durante las enfermedades moderada-grave, que alteran la ingesta de líquidos / salida, incluso si no tienen diagnóstico previo de diabetes insípida o hipernatremia.